

Labeling for
For STED microscopy, similar sample preparation techniques may be utilized as for conventional microscopy. However, the increase in special resolution requires additional precautions to ensure the structural preservation of the specimen.
STED microscopy
Since the discovery of the diffraction barrier of light microscopy in the late nineteenth century1, it has been accepted that conventional far-field optical microscopy cannot resolve structural details finer than half the wavelength of light. Superresolution fluorescence microscopy techniques, such as stimulated emission depletion (STED) microscopy2, 3, overcome this limitation. Here, we present robust sample reparation protocols for STED microscopy on mammalian cells. These protocols can be used as a starting point to adapt existing labeling strategies to the requirements of STED microscopy.
Materials for immunofluorescence and phalloidin labeling of mammalian cells
Forceps
For handling coverslips, forceps with very fine tips are recommended, e.g. Dumont forceps No. 5 (straight) or Dumont forceps No. 7 (bent).

Methanol, abs. (TOXIC!)
For fixation, methanol (abs.) should be chilled to -20 °C.
Formaldehyde solution. (TOXIC!)
A freshly prepared formaldehyde solution buffered to a neutral pH is used for fixation. This solution should be prepared using para-formaldehyde. After preparation it should not be used longer than 1–2 weeks (when stored at +4 °C). Alternatively, formaldehyde solutions may be frozen at -20 °C/-80 °C immediately after preparation. Then they might be stored and used for longer time periods. The formaldehyde concentration and fixation time needs to be determined for each antigen/primary antibody. Typical concentrations are 2 % – 8 %.
Extraction solution
Buffered solutions of detergents such as Tween 20, Triton X-100, Saponin or SDS are used as extraction solution.

Figure 2. Humid Chamber
For many applications, the following solutions are used: PBS + 0.1 % – 0.5 % Triton X-100
„Humid chamber“
Several suppliers offer humid chambers. Alternatively, large glass petri dishes may be used which need to be equipped with a tray for coverslips (e.g. a pipette tip holder from a pipette tip box).
Phosphate buffered saline (PBS, pH 7–7.5)
Prepare as follows:
| Compound | Concentration |
|---|---|
| NaCl | 137 nM |
| KCl | 2.7 nM |
| Na2HPO4 | 10 nM |
| KH2PO4 | 2 nM |
Blocking solution
For specific immunolabeling of cellular structures, unspecific labeling needs to be avoided. To this end, solutions containing various proteins that do not interfere with phalloidin or antibody reactions are applied, e.g. bovine serum albumin, gelatin, yeast extract, or milk (from milk powder).
For many applications, the following solution is used: PBS + 0.1 % Tween 20 + 1 % – 5 % bovine serum albumin (BSA). In the following, the PBS/BSA/Tween 20 blocking soulution will be refered to as PBT.
Primary Antibodies
A wide variety of primary antibodies are available and may be used for STED microscopy. Examples are included in Table 1.
Secondary Antibodies
For STED microscopy, abberior dyes coupled to secondary antibodies may be used. Examples are included in Table 2.
The standard dilution factor for Abberior secondary antibodies is 1:200.
Fluorophore labeled phalloidin (TOXIC!)
Typically, phalloidin is shipped freeze-dried, i.e. in solid form. Upon arrival it must be dissolved in methanol (abs.), DMF (water free) or DMSO (water free), see Note 1.
Depending on application, cell line etc., phalloidin is used in concentrations between 1 unit/ml and 2 units/ml in aqueous buffer.
Embedding/ mounting media
A variety of embedding media is available. For 3D STED microscopy we recommend non-hardening embedding media such as abberior Mount Liquid Antifade. For 2D STED microscopy we recommend the following embedding media: abberior Mount Solid Antifade; Mowiol/ DABCO; Prolong Gold, Prolong Diamond (Note 2).
Coverslips
For the use of 60x & 100x oil immersion objective lenses, glass coverslips with a thickness of ~170 µm should be used i.e. No 1.5 or No 1.5H.
Please DO NOT USE plastic coverclips or live cell chambers with plastic bottoms.
Furthermore, no coverslips with grids, gratings or similar should be used, because those structures might interfere with image generation and generate aberrations.
Slides
The only limitation for slides is that they need to fit to the sample holder of the microscope. Super Frost slides or similar slides are not required but may be used.
Plastic or glass petri dishes
For fixation, blocking, and washing of the samples, plastic or glass petri dishes may be used.
Pipettes
Standard micropipettes with different, adjustable volumes.
Labeling Protocols
Protocol I
Immunofluorescence-labeling of cultivated adherent mammalian cells – methanol fixiation
1. Cultivation of cells
Cells are typically seeded on cover slips for 12–36 h before labeling (Note 3, 4)
| Target | Source Species | Antibody specificity | Fixation |
|---|---|---|---|
| Nuclear Pore complex | mouse (monoclonal) | Nup153 (abcam, ab24700) | Methanol or 2% – 8% formaldehyde |
| ccTubulin | mouse (monoclonal) | wα-Tubulin (SIGMA, T6074) | Methanol |
Table 1. Commercial primary antibodies for the creation of test samples.
| Dye | Excitation Line | Detection | STED |
|---|---|---|---|
| abberior STAR GREEN | 488 nm | 500 nm – 550 nm | 595 nm |
| abberior STAR ORANGE | 561 nm | 570 nm – 630 nm | 775 nm |
| abberior STAR RED | 640 nm | 650 nm – 750 | 775 nm |
Table 2. abberior dyes frequently used for STED imaging.
2. Fixation and blocking
Sample is fixed for 5 min with ice-cold methanol (abs.) with cells facing upwards (Note 5, 6, 7, 8, 9, 10). Then, cover slips are washed with PBS (Note 11, 12, 13). Finally, unspecific binding sides are blocked with PBT for >15 min at RT, e.g. in a petri dish (Note 14).
3. Incubation with primary antibodies
Coverslips are removed from the petri dish and excess PBT is drained by placing the edge of the coverslip on a piece of tissue. Then, coverslips are placed in a humid chamber, covered with 25 µl to 100 µl (depending on the diameter of the coverslips) of diluted antibody solution (in PBT) (Note 15), and incubated for 1 h at RT.
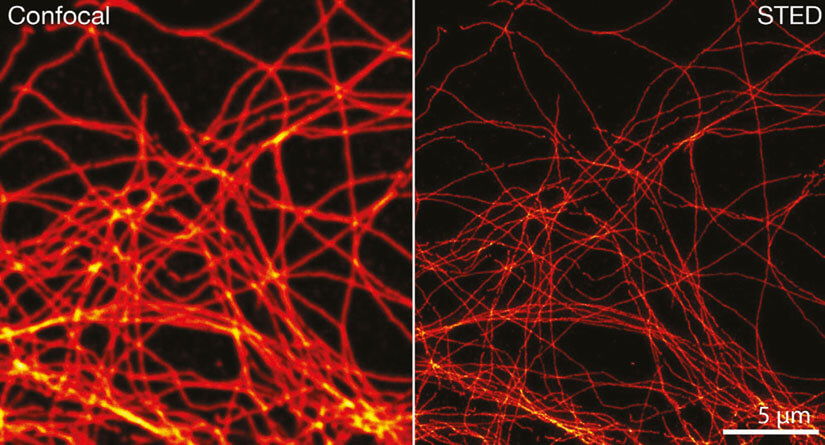
Figure 3. Confocal and STED image of methanol fixed cultivated mammalian cells labeled with tubulin specific primary antibodies and secondary antibodies coupled to abberior STAR RED.
4. Washing
The coverslips are removed from the humid chamber. The antibody solution is drained by pipetting off the liquid and then placing the edge of the coverslip on a piece of tissue. Then, the coverslips are placed in a fresh petri dish containing PBS (Note 13; 16) followed by incubation > 5 min at RT (2x). An additional blocking step with PBT is possible after this step (optional).
5. Incubation with secondary antibodies
Coverslips are removed from the petri dish and excess PBS is drained by placing the edge of the coverslip on a piece of tissue. Then, coverslips are placed in a humid chamber, covered with 25 µl to 100 µl (depending on the diameter of the coverslips) diluted antibody solution (in PBT) (Note 15), and are incubated for 1 h at RT.
6. Washing
The coverslips are removed from the humid chamber. Antibody solution is drained by pipetting off the liquid and then placing the edge of the coverslip on a piece of tissue. Then, coverslips are placed in a fresh petri dish containing PBS (Note 13, 16, 17). Incubate for at least 5 min at RT (3x).
7. Embedding, Storage, Stability
Finally, the coverslips are removed from the petri dish; excess PBS is drained by placing the edge of the coverslip on a piece of tissue. Then, the coverslips are mounted using the favored embedding medium.
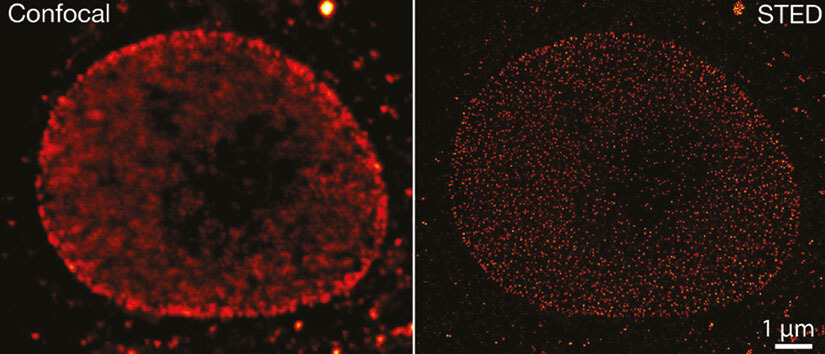
Figure 4. Confocal and STED image of formaldehyde fixed cultivated mammalian cells labeled with primary specific antibodies specific for Nup153 and secondary antibodies coupled to abberior STAR RED.
Protocol II
Immunofluorescence-labeling of cultivated adherent mammalian cells – formaldehyde fixiation
1. Cultivation of cells
The cells are typically seeded on cover slips for 12–36 h before labeling (Note 3, 4)
2. Fixation, Extraction and Blocking
Coverslips are fixed with formaldehyde solution with cells facing upwards (Note 5, 6, 7, 8, 9, 10) for 5 min. Then, cells are extracted using 0.1 – 0.5 % Triton X-100 in PBS (Note 11) also for 5 min. The coverslips are washed with PBS (Note 12, 13). Finally, unspecific binding sites are blocked with PBT for >15 min at RT (Note 14) in a petri dish.
3. Incubation with primary antibodies
Coverslips are removed from the petri dish; excess PBT is drained by placing the edge of the coverslip on a piece of tissue. Then, coverslips are placed in a humid chamber, covered with 25 µl to 100 µl (depending on the diameter of the coverslips) diluted antibody solution (in PBT) (Note 15), and are incubated for 1 h at RT.
4. Washing
The coverslips are removed from the humid chamber. The antibody solution is drained by pipetting off the liquid and then placing the edge of the coverslip on a piece of tissue. Then, the coverslips are placed in a fresh petri dish containing PBS (Note 13, 16). Incubation for at least 5 min at RT (2x). An additional blocking step with PBT is possible after this step (optional).
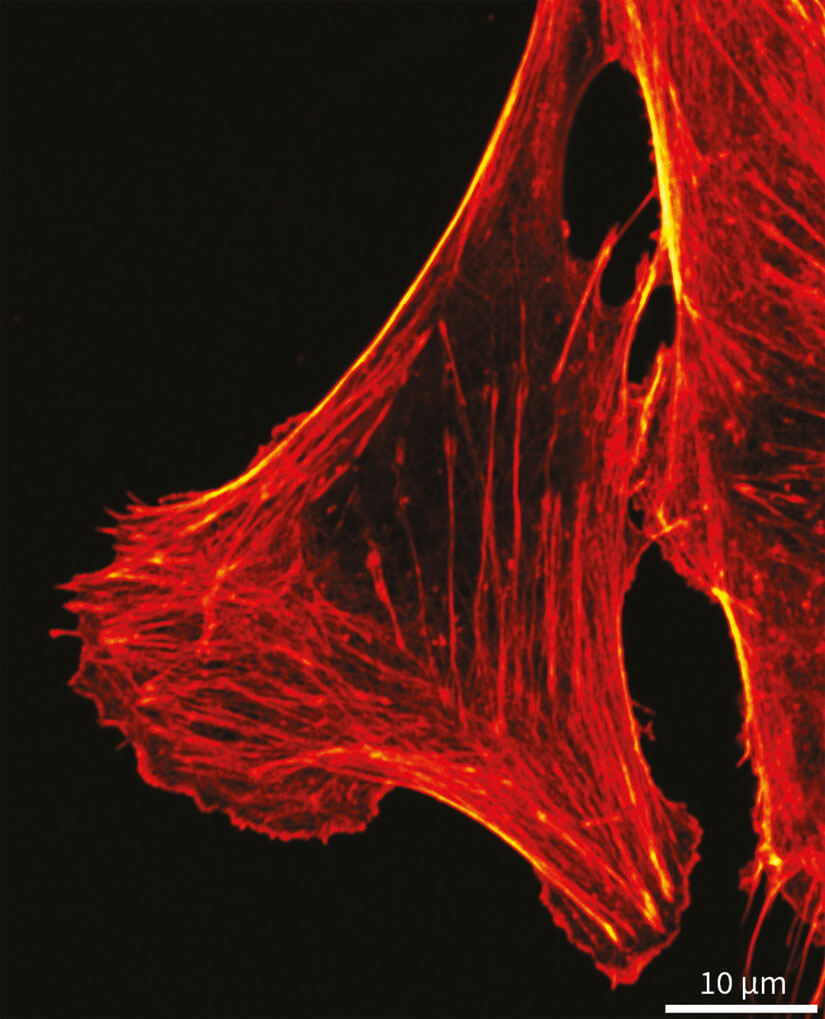
Figure 5. Confocal image of formaldehyde fixated cultivated mammalian cells labeled with phalloidin coupled to abberior STAR RED.
5. Incubation with secondary antibodies
Coverslips are removed from the petri dish; excess PBS is drained by placing the edge of the coverslip on a piece of tissue. Then, coverslips are placed in a humid chamber, covered with 25 µl to 100 µl (depending on the diameter of the coverslips) diluted antibody solution (in PBT) (Note 15), and are incubated for 1 h at RT.
6. Washing
The coverslips are removed from the humid chamber. The antibody solution is drained by pipetting off the liquid and then placing the edge of the coverslip on a piece of tissue. Then, the coverslips are placed in a fresh petri dish containing PBS (Note 13, 16, 17). Incubation for at least 5 min at RT (3x).
7. Embedding, Storage, Stability
Finally, the coverslips are removed from the petri dish; excess PBS is drained by placing the edge of the coverslip on a piece of tissue. Then, the coverslips are mounted using the favoured embedding Medium. In addition, mounted coverslips may be fixed with nail polish completely or at several small points (Note 18, 19).
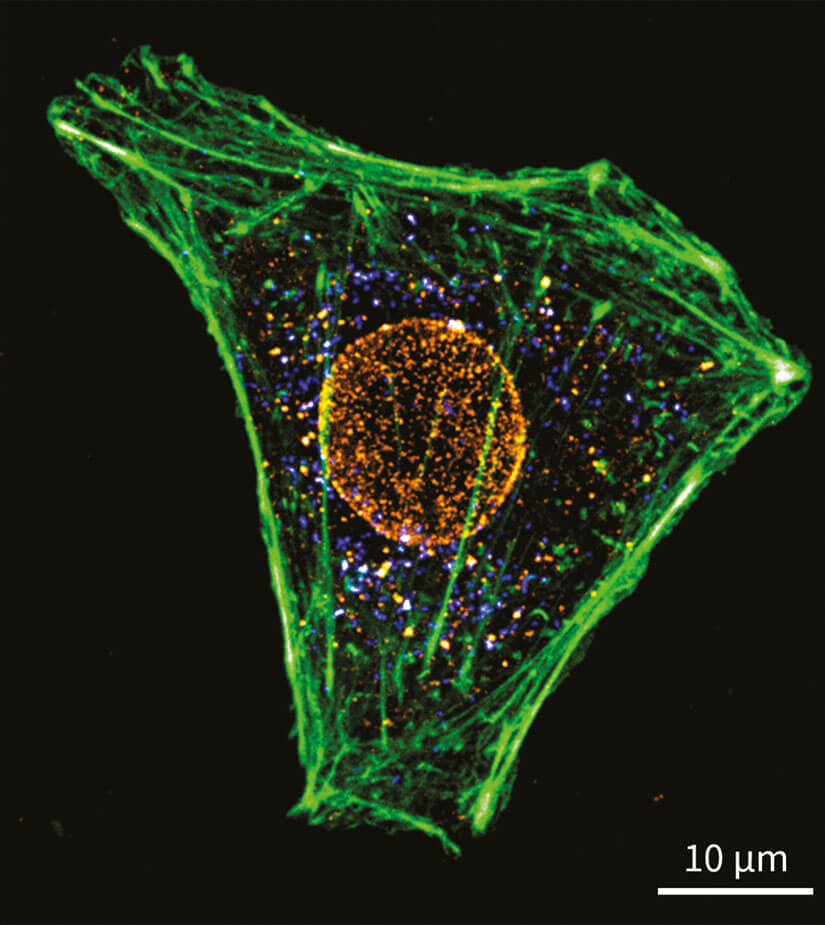
Figure 6. Three-color confocal image of formaldehyde fixed cultivated mammalian cells labeled for phalloidin with abberior STAR GREEN together with immuno-labeled nuclear pore complex (Nup153, abberior STAR 580) and peroxisomes (PMP70, abberior STAR RED.)
8. Storage of samples
Ready-made samples should be stored at 4 °C (Note 21). Most samples are stable for a rather short time only. For best results (Fig. 5), samples should be imaged within one week.
Protocol III
Phalloidin-labeling of cultivated adherent mammalian cells – Fformaldehyde fixiation
1. Cultivation of cells
The cells are typically seeded on coverslips for 12–36 h before labeling (Note 3; 4)
2. Fixation, Extraction and Blocking
Samples are fixed with formaldehyde solution with cells facing upwards (Note 5, 6, 7, 8, 9, 10) for 5 min. Then cells are extracted using 0.1–0.5 % Triton X-100 in PBS (Note 11) for 5 min. The coverslips are washed with PBS (Note 12, 13). Finally, unspecific binding sides are blocked with PBT for >15 min at RT (Note 14) in a petri dish.
3. Incubation with fluorophore-labeled phalloidin
Coverslips are removed from the petri dish; excess PBT is drained by placing the edge of the coverslip on a piece of tissue. Then, coverslips are placed in a humid chamber, covered with 25 µl to 100 µl (depending
on the diameter of the coverslips) diluted phalloidin solution (in PBT; Note 15), and are incubated for 1 h at RT.
4. Washing
The coverslips are removed from the humid chamber. The antibody solution is drained by pipetting off the liquid and then placing the edge of the coverslip on a piece of tissue. Then, the coverslips are placed in a fresh petri dish containing PBS (Note 13, 16). Incubation for at least 5 min at RT (2x).
5. Embedding, Storage, Stability
Finally, the coverslips are removed from the petri dish; excess PBS is drained by placing the edge of the coverslip on a piece of tissue. Then, the coverslips are mounted using the favoured embedding medium. In addition, mounted coverslips may be fixed with nail polish completely or at several small points (Note 18, 19).
6. Storage of samples
Ready-made samples should be stored at 4 °C (Note 21). Most samples are stable for a rather short time only. For best results (Figs. 5, 6), samples should be imaged after not much more than one week.
Notes
The most important rule for immuno-labeling is that specimens must not be allowed to dry out.
- Dissolution of fluorphore-labeled phalloidin: The vial contents should be dissolved to yield a final concentration of 200 units/ml, which is equivalent to approximately 6.6 µM.
- Certain embedding media may affect the photophysical properties of some fluorophores, leading to changes in brightness and bleaching behaviour, for example. A shift in the excitation and emission spectra is also possible.
- Seeding of cells may take place earlier if required. Depending on the doubling time, cultivation time, cell density and others, cells may grow in microcolonies.
- Cells grown in very high densities, i.e. a confluent layer, may lead to high background signal during imaging.
- In contrast to other protocols, specimen/cells are not washed with buffer or similar before fixation.
- Cells are not subjected to a pre-extraction procedure before fixation.
- The fixation is performed using an excess of fixative.
- For fixation, the coverslips are placed into a fresh petri dish containing fixative. Alternatively, fixation may be performed in the same petri dish the cells were grown in.
- For fixation, the growth medium is removed completely. Then, cells are submerged in fixative.
- The fixation should be performed with freshly prepared or frozen fixative. It is best to use prewarmed fixative. Ready made 37% formaldehyde/ methanol should not be used.
- Between fixation and blocking, an extraction step may be required. For formaldehyde fixation it is crucial to extract the cells using detergents. For methanol fixed cells, extraction is not required, however a short wash in PBS/ 0,1% Triton X-100 may be advantageous for the labeling.
- Coverslips with fixed cells may be stored several (1–2) days at 4 °C in PBS. However, the quality of the labeling may be affected by the storage of the samples.
- Washing should be performed using excess PBS.
- Blocking should be performed using excess PBT solution.
- For antibody incubation, coverslips should not be placed with cells facing downward on parafilm or similar films. Although this might reduce the amount of antibody needed for labeling, cells may be affected while removing them from the parafilm.
- After antibody incubation, coverslips should be washed in different petri dishes. Otherwise cross contaminations might occur.
- If high background labeling of the specimen occurs, it can be further reduced by incubation with PBS/ Triton X-100 and/or PBT.
- If non-hardening embedding media are used, coverslips have to be sealed using Twinsil or nail polish.
- If TDE is used as embedding medium, twinsil may not be used since the TDE will hinder its polymerization and hardening.
- The use of TDE is not possible for phalloidin labeled cells.
- Storage of samples at -20 °C should be avoided, because ice crystals may affect the quality of the samples.
Abbreviations
BSA
DMF
DMSO
PBS
PBT
RT
SDS
STED
TDE
Bovine Serum Albumin
N,N-Dimethylformami
Dimethylsulfoxide
Phosphate buffered saline
PBS/ BSA/ Tween 20
Room Temperature
Sodium Dodecyl Sulfate
Stimulated Emission Depletion
2,2′-Thiodiethanol
1 “Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung”. Abbe, E. (1873): Arch. Mikr. Anat. 9: 413–420.
2 “Breaking the diffraction resolution limit by stimulated emission: stimulated emission depletion microscopy”. Hell, S. W., & Wichmann, J. (1994): Opt. Lett. 19: 780–782
3 “Fluorescence nanoscopy in cell biology”. Sahl, S. J. , S. W. Hell, S. Jakobs (2017): Nature Rev. Mol. Cell Biol. 18, 685-701.
4 “Sample Preparation for STED Microscopy”. Wurm, C. A. , D. Neumann, R. Schmidt, A. Egner, S. Jakobs (2010): Methods Mol. Biol. 591, 185 – 199.