

Let the cells shine
Coarse cellular structures can sufficiently alter passing light such that they are directly visible through a light microscope. For visualizing smaller structures, light microscopy relies on specific labeling with fluorescent dyes. The dye allows to visualize target molecules indirectly by observing its fluorescence emission.
with immunofluorescence labeling
A precise and powerful tool
Naturally, a normal cell or tissue sample does not come along with fluorophores, meaning that you have to introduce them into the specimen before acquiring images. There are different strategies to bring the dye to the sample, e.g. co-expression of a fluorescent protein or click chemistry. The most versatile and therefore most common strategy, however, is immunofluorescence. In case you always wanted to know how immunofluorescence works and which properties of antibodies make it so powerful and at the same time define its limits, read on!
In immunofluorescence labeling, the fluorophore is brought to the biomolecule of interest, usually a protein, with the help of antibodies. And since antibodies play such a central role here, it is worth to take a closer look at them before diving into the technical aspects of immunofluorescence.
Antibodies or immunoglobulins are an integral part of the immune system. They are produced by specialized immune cells, the B-cells, when these encounter a foreign molecule, e.g. some part of a virus or bacterium. The antibodies help the immune system to recognize and fight this molecule and the pathogen it belongs to. Two properties make antibodies perfectly suited for this task: specificity and affinity, meaning that they are particularly good at finding exactly their counterpart, and once they have bound to it they never let go again. For these reasons, antibodies are great not only for our immune system to identify and label intruders, but also in biological research to detect, mark, or isolate proteins of interest.
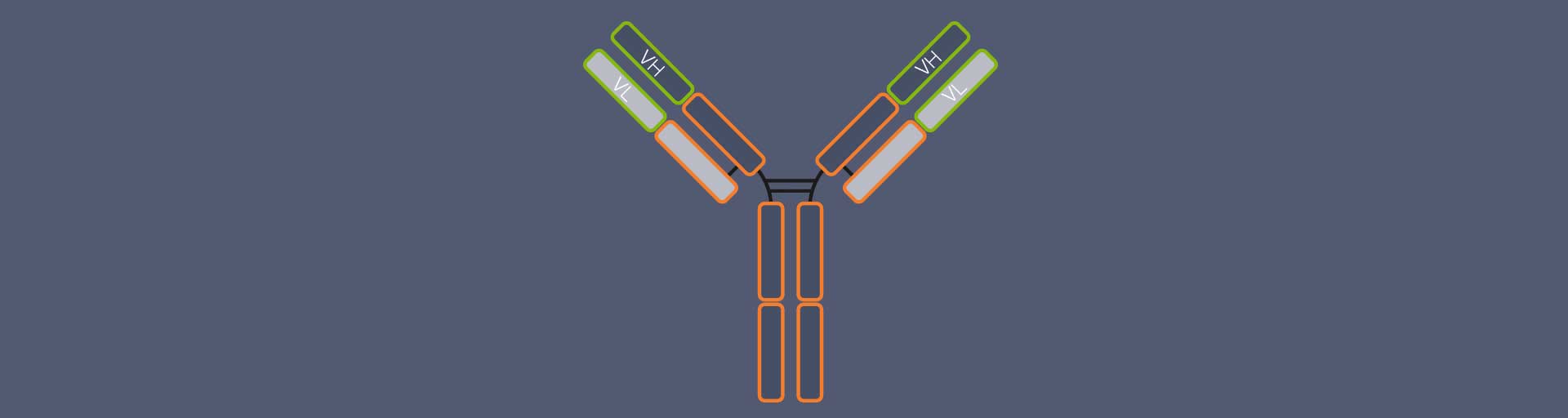
Most people will probably recognize an antibody depiction when they see one, since it is so familiar: Antibodies are Y-shaped proteins, formed by four chains: two so-called heavy chains and two light chains (Fig. 1). The antibody’s stem and part of the arms are identical for all antibodies of one class – e.g. immunoglobulin type G (igG), the most common antibody – and are therefore together called the constant region. The outermost part of the arms, however, differs from antibody to antibody. It is called the variable region. The variable region of the heavy chain (VH) and the variable region of the light chain (VL) together form the antigen binding domain. It is this part of the antibody which recognizes and binds the target structure (“epitope”), hence determining the antibody’s specificity.1
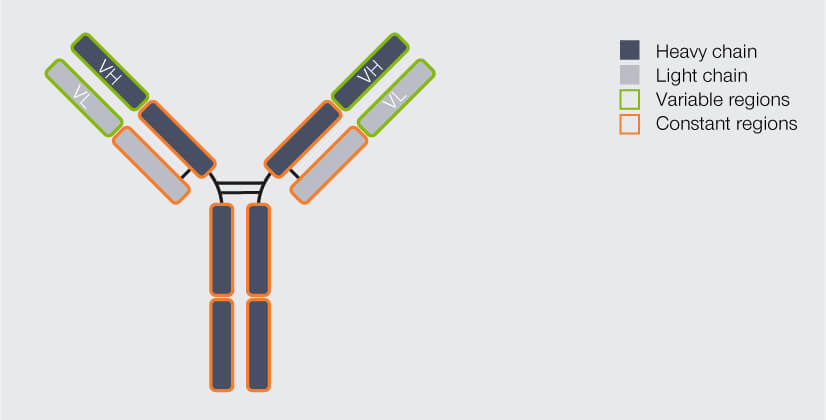
Figure 1. Structure of an IgG antibody (AB). The AB consists of four chains, two heavy and two light ones. The chains’ regions at the end of the arms differ from AB to AB, they are variable. The variable region of the heavy chain (VH) and of the light chain (VL) together form the antigen binding domain, which determines which epitope the AB recognizes and binds to. The rest of the AB is identical for all IgGs and called the constant region.
For scientific applications, antibodies are produced in animals, mostly mice, rats, rabbits, or bigger animals like donkeys and goats. Naturally, an animal’s B-cells react to a foreign molecule by starting mass assembly of antibodies for tagging it. We can hijack this immune response simply by injecting an animal with a protein of interest, whereupon the animal’s B-cells will happily produce antibodies specifically against this protein. Exactly what we need!
An important differentiation for antibodies used in immunofluorescence is their clonality: They can either be mono- or polyclonal (Fig. 2). The antibodies produced by an individual B-cell are all absolutely identical, meaning that they are of the same type (e.g. IgG) and all recognize and bind to the same epitope.
When a pool of antibodies is produced by clones of this B-cell, they are said to be monoclonal, meaning all of them recognize the same epitope. Imagine having several keys for the same building, which are exactly the same and which all open the exact same lock. The second option are antibodies which recognize the same protein, but different epitopes of it, because they originate from a pool of different B-cells. These antibodies are called polyclonal. This would be akin to having keys for the same building (as in “same protein”), but they all “recognize” different locks on different doors.
If you know that someone has a “monoclonal” key, you know exactly which door he used to get in as the key will fit in this door’s lock only. Similarly, monoclonal antibodies exhibit superior specificity and very low cross-reactivity, i.e. they are unlikely to bind to anything other than their epitope. In immunofluorescence, this results in low background noise. However, they give rather low signal, since only one antibody can bind per molecule, and are more expensive to produce. Moreover, as they are all the same, when attaching a fluorescent label happens to change their binding behavior, it will do so for all antibodies, meaning they will all show decreased functionality.
In contrast, someone with a “polyclonal” key set has more flexibility as to which door she uses, but also, you can’t be so sure which one it is. Correspondingly, polyclonal antibodies are comparably inexpensive, exhibit a higher signal as they can bind more than one epitope of the target protein, and tagging with a fluorophore is unlikely to affect the binding behavior of all of them. On the other hand, the properties and quality of polyclonal antibodies may vary between badges and they are more likely to cross-react with proteins other than the one of interest, producing more background noise.1
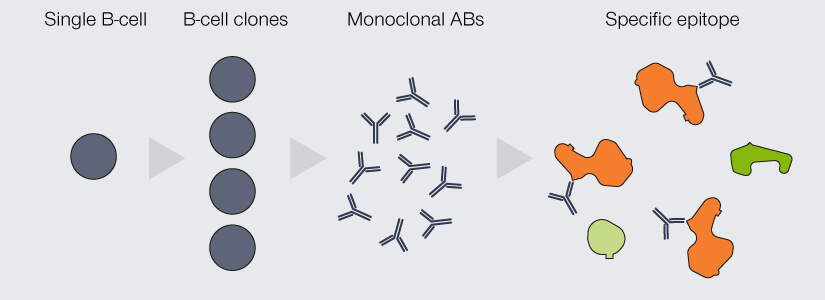
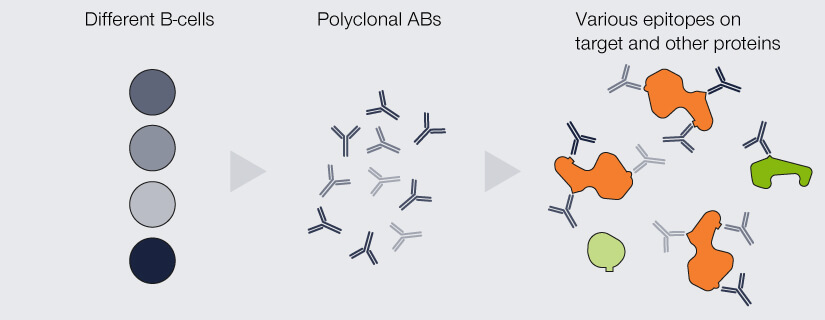
Figure 2. Differences between monoclonal and polyclonal antibodies (ABs). Monoclonal ABs are produced by the clones of a single B-cell. They are completely identical and all recognize and bind the same epitope. Due to their specificity, they display very low cross-reactivity but are comparably insensitive as every target protein is bound by only one AB. Polyclonal ABs originate from a pool of B-cells. They recognize a variety of epitopes on the target protein, which increases sensitivity, but might bind to epitopes of other proteins than the one of interest.
Two steps for a better signal
With this information in mind, let’s go back to immunofluorescence staining. There are two different labeling strategies (Fig. 3): Direct immunofluorescence staining uses a single antibody which binds to the protein of interest and carries a fluorophore. Indirect immunofluorescence staining, in contrast, requires two antibodies that are applied sequentially: Like in direct immunofluorescence staining, the so-called primary antibody binds to the protein of interest. It mostly originates from mice or rabbits. However, it does not carry the fluorophore. This is introduced by the secondary antibody, which is usually produced in donkey, goat, or rabbit by immunizing these animals with antibodies from mouse or rabbit. The primary antibody binds to the constant region of the primary antibody.
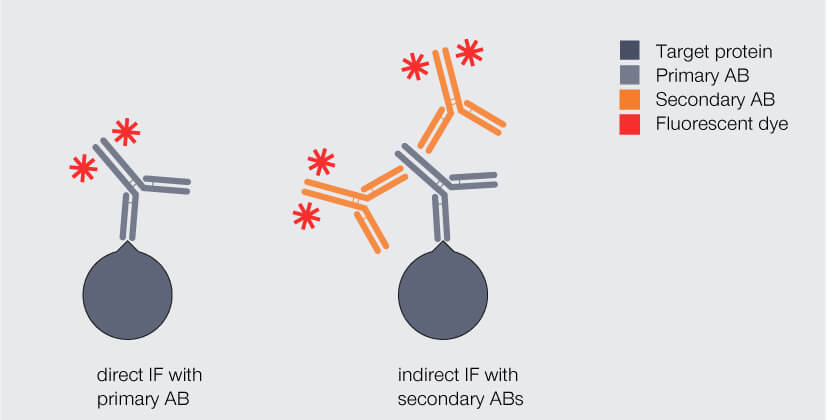
Figure 3. Direct and indirect immunofluorescence (IF) staining. Dark gray: target protein, light gray: primary antibody (AB), orange: fluorophore-coupled AB.
At first glance, direct IF labeling appears much more attractive as it offers an easy and fast staining protocol. Why not go for it? There are two main drawbacks: First, the number of commercially available fluorophore-coupled primary antibodies is limited as for every protein an individual antibody has to be produced and coupled to a fluorophore. And chances are that none of the antibodies available is directed against your protein of interest. Second, in direct immunofluorescence labeling any target protein will be bound by maximum one fluorophore-coupled antibody and will therefore produce a comparably weak fluorescent signal.
For these reasons, indirect immunofluorescence labeling is usually the strategy of choice. Here, two monoclonal or even more polyclonal antibodies carrying fluorophores bind to a single primary antibody, which amplifies the signal and boosts sensitivity. 2,3 This way, the primary antibody can focus on recognizing a very specific target of interest, while the secondary antibody merely has to be targeted against the primary antibody.
Knowing the limits
With indirect immunofluorescence researchers have a particularly versatile technique at hand to label proteins with high specificity and sensitivity. The most important advantage when using antibodies in immunofluorescence is that you can target virtually any protein as long as there is an antibody at hand that recognizes this target.
So much for the advantages. But every method has its limits, doesn’t it? Indeed this is also the case for immunofluorescence. For one thing, the number of different proteins one might label in a single sample is restricted, because for every primary antibody used you need a different host species. And, as primary antibodies are usually generated in rabbits and mice, it is usually not possible to stain more than two proteins at once: the secondary antibody would not be able to differentiate between e.g. two mouse antibodies bound to two different target proteins and you would consequently have no chance to label these two targets with different fluorophores.
For another thing, coming in at around 30 nm, the size of the complex of primary and secondary antibodies might pose a problem: In the tightly packed environment of a cell, the complexes might block each other’s access to epitopes. Moreover, their size means that the fluorophore might be displaced from the labeled protein by something like 20 nm. As long as resolution in light microscopy was restricted to the Abbé limit of about 250 nm, this so-called linkage error was negligible. With the invention of superresolution microscopy, however, 20 nm offset between target and label mean that otherwise sharp images get blurred.
Luckily, a new type of antibody provides a solution for both problems. But this is the topic of another article.
1 Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR J. 2005;46(3):258-68. doi: 10.1093/ilar.46.3.258.
2 Im K, Mareninov S, Diaz MFP, Yong WH. An Introduction to Performing Immunofluorescence Staining. Methods Mol Biol. 2019;1897:299-311. doi: 10.1007/978-1-4939-8935-5_26.
3 Carrington G, Tomlinson D, Peckham M. Exploiting nanobodies and Affimers for superresolution imaging in light microscopy. Mol Biol Cell. 2019 Oct 15;30(22):2737-2740. doi: 10.1091/mbc.E18-11-0694.