Why do superresolution microscopists love alpacas?
It is a very simple yet very important fact: that the protein localization precision of any superresolution microscope can only be as good as the size of the fluorescent staining allows. In other words, when your fluorescent dye is too big or too far away from the protein you want to label, the resolving power of your microscope can be as high as anything – you will always be left with some uncertainty as to where exactly your protein is located. The good news is: there are ways to reduce the offset between target protein and fluorescent label. And one of these are nanobodies.
Because of their nanobodies!
Nanobodies for superresolution microscopy
In our knowledgebase you can read a lot about “How the donut changed the world” and the different ways how resolution beyond the diffraction limit can be achieved in fluorescence microscopy, with MINFLUX capable of resolving individual molecules. We talk a lot about optical physics, mathematical functions, and stuff like this. Which is all good and true.
However, what we have not touched on so far is the difference between resolution and localization precision: Superresolution microscopy achieves a resolution in the nanometer range. This resolution, however, applies only to the detected fluorescent molecules. A resolving power of 3 nanometers does not necessarily mean that the proteins labeled with the fluorophores can be localized with equal precision. When your fluorescent dye is too big or too far away from your protein of interest, you will never be able to localize it with a precision that is higher than this offset. But there are ways to reduce the offset between target protein and fluorescent label. And one of these are nanobodies, also called VHH fragments or single-domain antibodies.
Nanobodies are particularly small and simple-built antibodies derived from certain types of antibodies only found in camelids, a group of animals including camels, llamas, and alpacas. To understand the advantages of using nanobodies in superresolution microscopy, we first need to shortly recap how immunofluorescence staining works. For a more detailed introduction to the topic, check out our article “Let the cells shine with immunofluorescence labeling”.
Bringing the label to the target
In immunofluorescence staining, the biomolecule of interest – usually a protein – is labeled with a fluorophore with the help of antibodies. Typically, antibodies of immunoglobulin type G (IgG) are used. There are two different labeling strategies (Fig. 1): Direct immunofluorescence staining uses a single antibody which binds to the protein of interest and carries a fluorophore. Indirect immunofluorescence staining, in contrast, requires two antibodies that are applied sequentially: Like in direct immunofluorescence staining, the so-called primary antibody binds to the protein of interest. However, it does not carry the fluorophore. This is introduced by the secondary antibody which binds to the primary antibody.
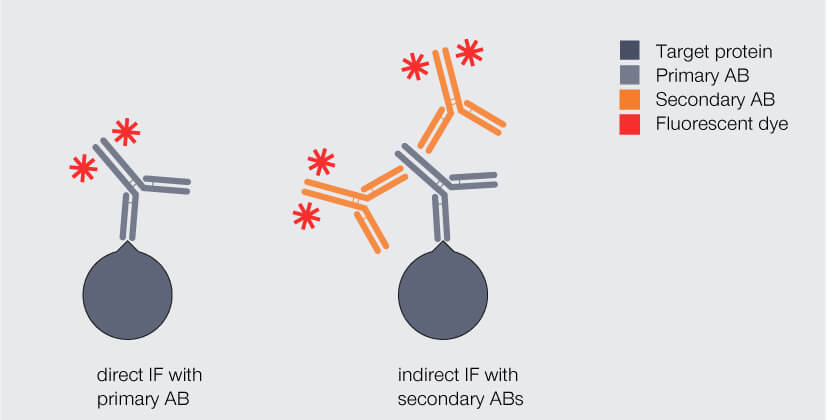
Figure 1. Direct and indirect immunofluorescence (IF) staining with antibodies (AB). Dark gray: target protein, light gray: primary antibody, orange: fluorophore-coupled antibody.
In most settings, indirect immunofluorescence labeling will be the strategy of choice as it has some advantages over direct immunofluorescence labeling. One is a brighter fluorescence signal when multiple, mostly polyclonal (what’s that again?) secondary antibodies carrying fluorophores bind to a single primary antibody, boosting sensitivity.1,2
So with indirect immunofluorescence labeling, we get a brighter signal, which is good. However, with indirect immunofluorescence labeling we also get a molecular complex of primary and secondary antibodies with a proud size of 30 nm in diameter, which is not so good: It means that the fluorophore may be something like 20 nm away from the protein. That is the so-called linkage error. It is no issue as long as you are imaging with a conventional confocal microscope with a maximum resolution of roughly 250 nm, where less than ten percent linkage error hardly matters. Superresolution microscopy, in contrast, resolves details of 20 to 30 nm in the case of STED or PALM/STORM; with MINFLUX, we even talk about 2 to 3 nm!
Which brings us back to the initial problem: In superresolution microscopy, a big antibody complex means a significant offset between target and label, limiting the achievable localization precision. Another drawback of such large labeling complexes is that they might block each other’s access to the protein targets when these are densely packed – which is often the case in the busy and complex environment of cells or tissues.3,4,5
Nanobodies for the nanoscale
By now, you probably guessed how nanobodies can help here. It’s all in their name: Compared to conventional IgG antibodies, which measure 12 nm in length, nanobodies are tiny at 3 nm. The difference becomes even more obvious when we look at the molecular weight (this is a measure for the mass of a molecule, given in the quantity Dalton): An IgG antibody (150 kDa) outweighs a nanobody (12 to 15 kDa) by a factor of 10.2
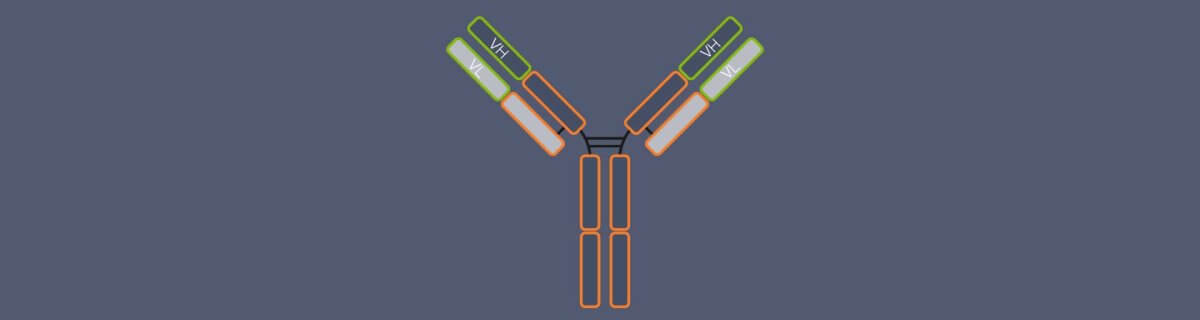
Nanobodies are so small because practically they are just fragments of the camelid antibodies, which already have a simpler structure than conventional antibodies: Conventional antibodies consist of four peptide chains, two heavy and two light ones, and the region that binds the target structure – the antigen-binding domain – is composed of two chain regions, the variable regions of the heavy chain (VH) and of the light chain (VL). Camelid antibodies lack the light chains; consequently, their antigen-binding domain consists of a single region only, called variable heavy region of the heavy chain (VHH). Nanobodies are derived from this VHH (Fig. 2).2 So, in fact, a nanobody is an antibody reduced to the essentials.
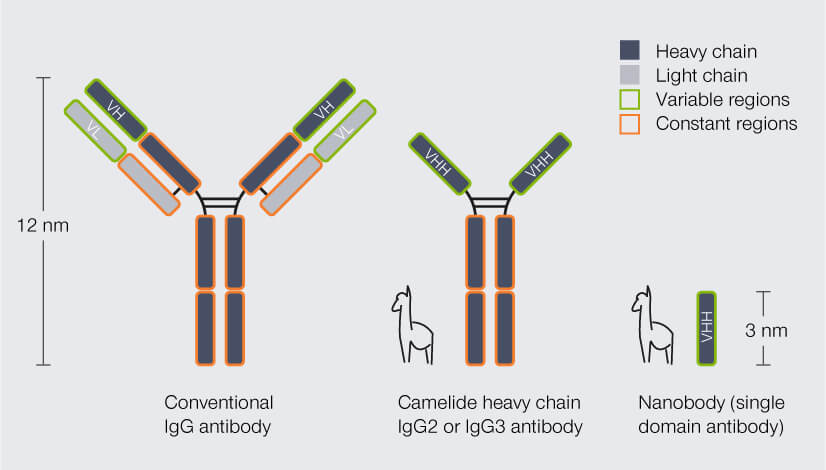
Figure 2. Structure and size of conventional IgG antibodies, heavy chain antibodies from camelid species (e.g. alpaca), and nanobodies. In conventional antibodies, the antigen-binding domain is formed by the variable region of the heavy chain (VH) and the variable region of the light chain (VL). Heavy chain camelid antibodies lack the light chains and their antigen-binding domain comprises the variable heavy region of the heavy chain (VHH) only. Nanobodies are derived from the VHH. In contrast to conventional and camelid antibodies, they do not have a constant region.
Due to their small size, nanobodies can significantly reduce the linkage error. But how much? And how can they be employed best? One option is to replace the fluorophore-coupled secondary antibody with a polyclonal secondary nanobody. This nanobody can bind to multiple sites of the primary antibody, including epitopes that cannot be recognized by conventional secondary antibodies. As a result, signal amplification remains high. The complex of primary antibody and secondary nanobodies is only slightly larger than a single antibody (Fig. 3).
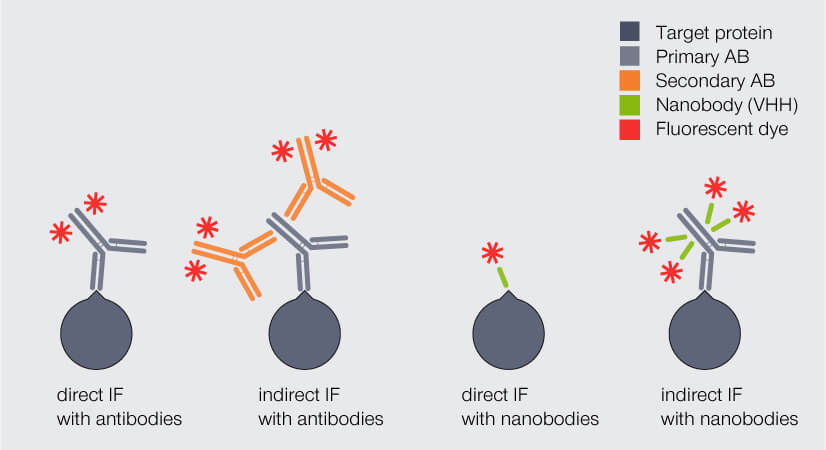
Figure 3. Direct and indirect immunofluorescence (IF) staining with antibodies (AB) and nanobodies, respectively. The complex of primary AB and secondary nanobodies is only slightly larger than a single antibody and significantly smaller than the complex of primary and secondary AB.
Thus, with polyclonal secondary nanobodies, the linkage error is reduced while strong fluorescence signal is maintained. For instance, tubulin fragments stained with secondary nanobodies coupled to abberior STAR RED have a significantly smaller apparent filament diameter (67 nm) than tubulin filaments stained with secondary antibodies (77 nm) (Fig. 4). The linkage error can be reduced even further when fluorophore-coupled nanobodies are used for direct immunofluorescence. However, here the same limitations apply as for direct immunofluorescence with conventional antibodies: no signal amplification and a limited availability of suitable nanobodies.
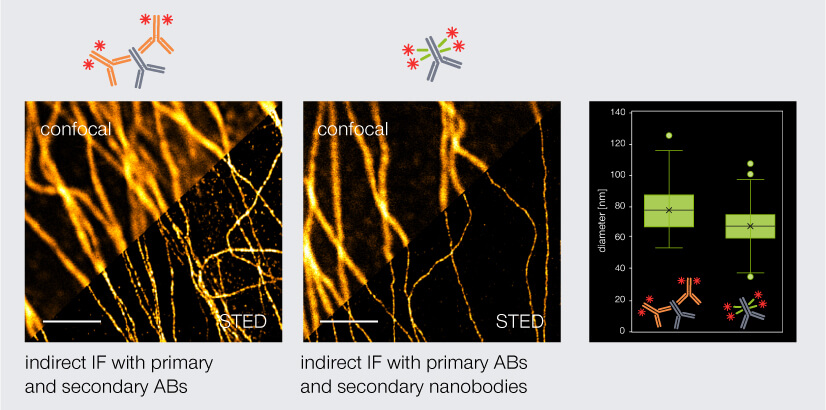
Figure 4. Confocal and STED images of tubulin stained via indirect immunofluorescence. Primary mouse anti-tubulin antibodies (AB) were targeted either by secondary anti-mouse antibodies (left) or secondary anti-mouse nanobodies (middle). Secondary nanobodies reduce the linkage error and thus the apparent diameter of tubulin by 10 nm. Scale bars: 2 µm.
Multiplex staining: more colors for your image
Increasing resolution can open up opportunities unavailable to biological research before, as we discuss in “Superresolution for biology: when space, time, and context matter”. Another screw one can adjust in fluorescence microscopy to get more information out of ones image is the number of different proteins labeled in a single sample. Here, as well, a lot depends on the antibodies used.
As detailed above, indirect immunofluorescence staining is usually a two-step process where the sample is first incubated with the primary antibody and subsequently with the secondary antibody. This limits the number of target proteins that can be labeled since every primary antibody has to originate from a different host species to exclude cross-reactivity with the secondary antibodies. In theory, this cross-reactivity could be avoided by pre-incubating every individual primary antibody with its specific secondary antibody before adding the pre-assembled complex to the sample. However, this approach is not feasible when using conventional secondary antibodies as these are polyvalent (not to be confused with polyclonal), meaning that every antibody has two antigen-binding domains (one at each arm’s end). Consequently, it can bind to two target primary antibodies, which is also what happens in case you pre-incubate the two together. And as not only one, but many secondary antibodies do so, this results in the formation of huge antibody aggregates that are no longer able to deliver a specific immunostain.
Again, nanobodies are the solution, because they differ from conventional antibodies not only in size, but also in structure: As nanobodies consist of a single antigen-binding domain, they bind only a single target, i.e. they are monovalent and can easily be pre-mixed with primary antibodies (Fig. 5). Et voilá, antibody aggregates are history.
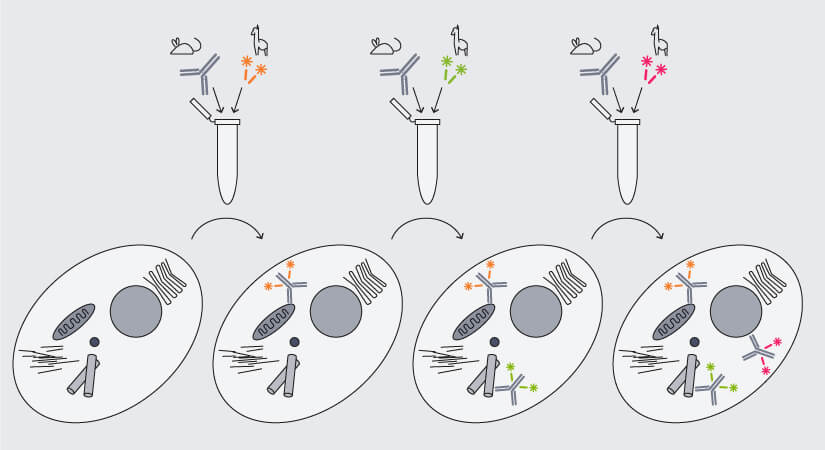
Figure 5. Multiplex immunofluorescence staining with secondary nanobodies following a premix&stain protocol. Primary antibodies against different targets raised in the same species are each premixed with secondary nanobodies coupled to different dyes and sequentially added to the sample.
With secondary nanobodies, it is now possible to use several primary antibodies raised in the same species to label a variety of target proteins, a strategy called multiplex staining.3 A 3-color STED image with excellent multicolor staining is shown in figure 6.
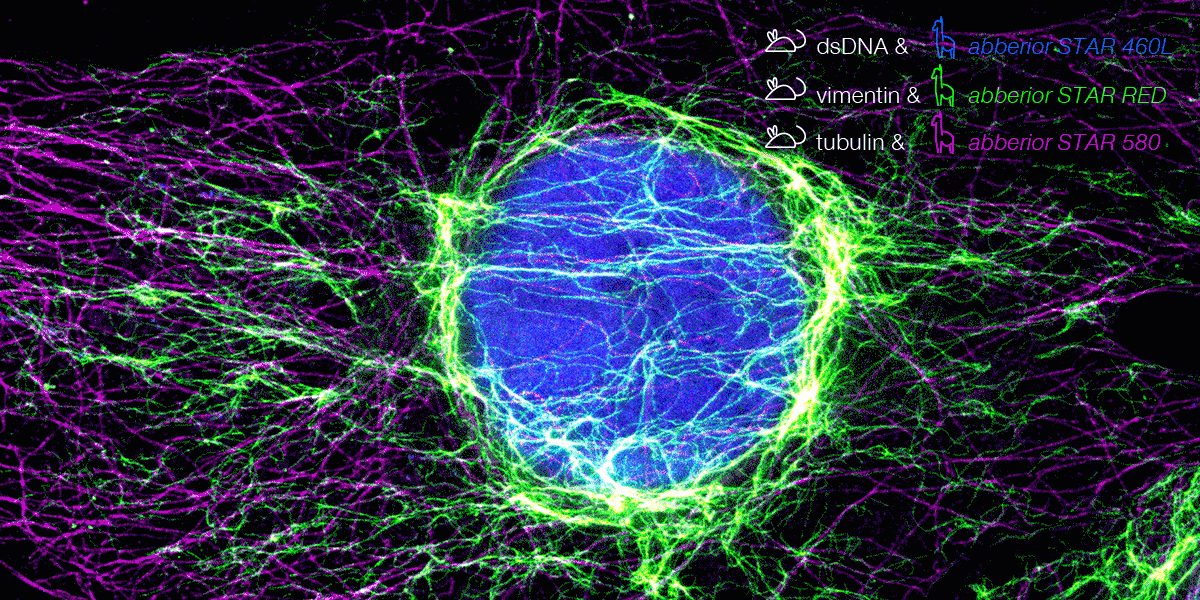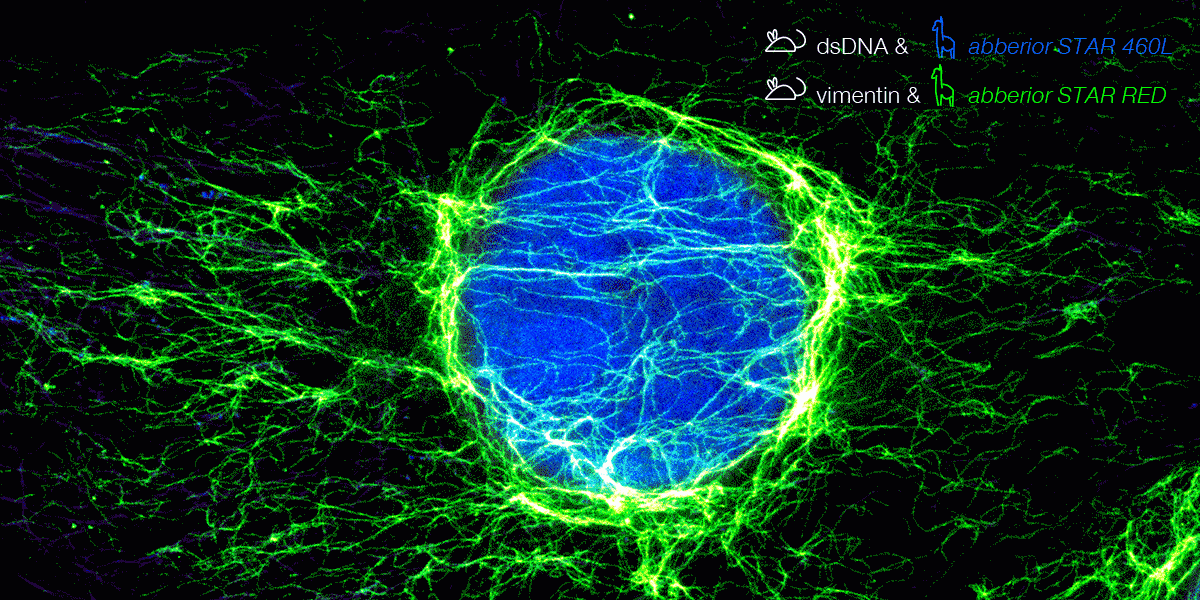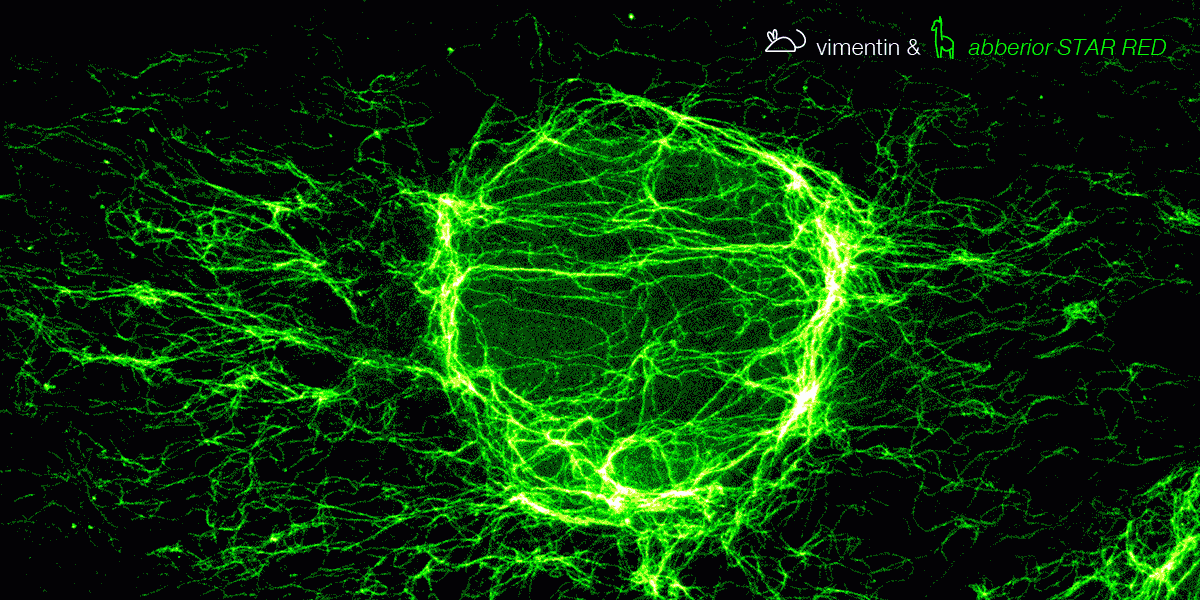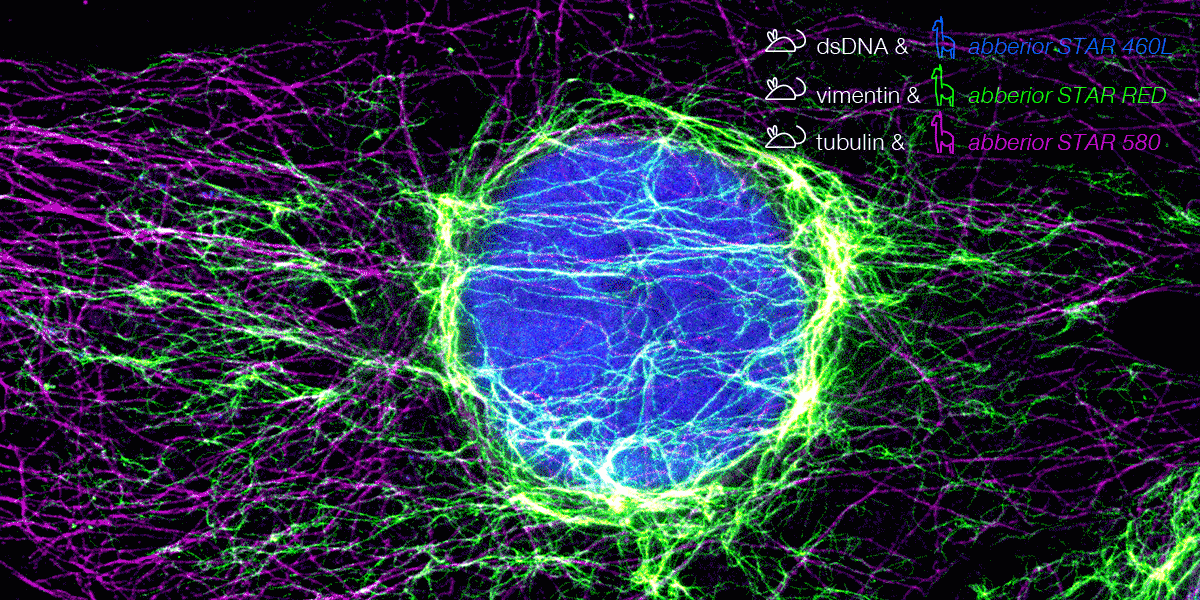
Figure 6. 3-color STED image stained via indirect IF with premixed complexes from primary antibodies raised in mouse and secondary nanobodies coupled to dyes. Stained structures are vimentin (green), tubulin (magenta) and dsDNA (blue).
Nanobodies have quickly developed into versatile tools in molecular biology. For fluorescence microscopy, they already solve more than one problem. And there is more to come, for sure.
1 Im K, Mareninov S, Diaz MFP, Yong WH. An Introduction to Performing Immunofluorescence Staining. Methods Mol Biol. 2019;1897:299-311. doi: 10.1007/978-1-4939-8935-5_26.
2 Carrington G, Tomlinson D, Peckham M. Exploiting nanobodies and Affimers for superresolution imaging in light microscopy. Mol Biol Cell. 2019 Oct 15;30(22):2737-2740. doi: 10.1091/mbc.E18-11-0694.
3 Sograte-Idrissi S, Schlichthaerle T, Duque-Afonso CJ, Alevra M, Strauss S, Moser T, Jungmann R, Rizzoli SO, Opazo F. Circumvention of common labelling artefacts using secondary nanobodies. Nanoscale. 2020 May 14;12(18):10226-10239. doi: 10.1039/d0nr00227e.
4 Ries J, Kaplan C, Platonova E, Eghlidi H, Ewers H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat Methods. 2012 Jun;9(6):582-4. doi: 10.1038/nmeth.1991. Epub 2012 Apr 29.
5 Gomes de Castro MA, Höbartner C, Opazo F. Aptamers provide superior stainings of cellular receptors studied under super-resolution microscopy. PLoS One. 2017 Feb 24;12(2):e0173050. doi: 10.1371/journal.pone.0173050.
6 Pleiner T, Bates M, Görlich D. A toolbox of anti-mouse and anti-rabbit IgG secondary nanobodies. J Cell Biol. 2018 Mar 5;217(3):1143-1154. doi: 10.1083/jcb.201709115. Epub 2017 Dec 20.