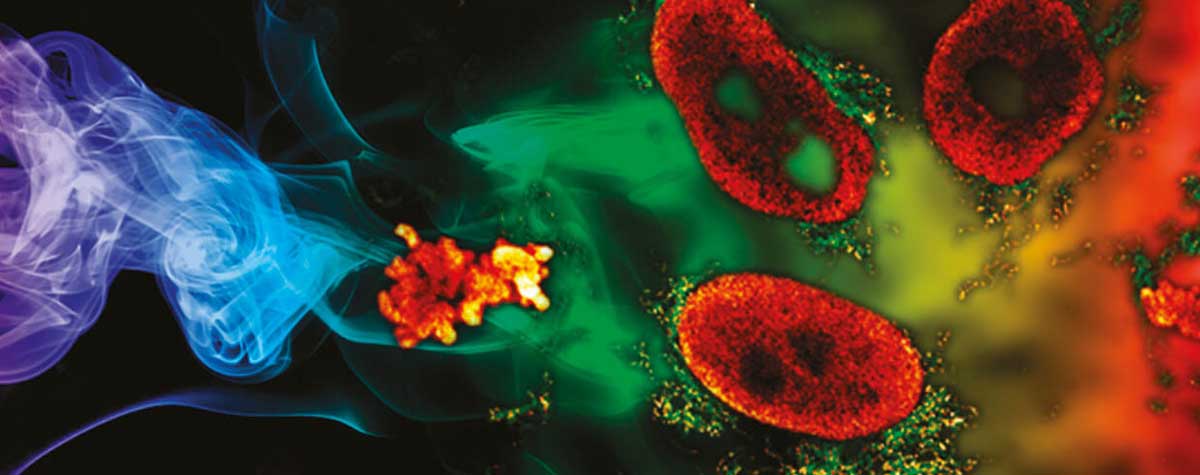
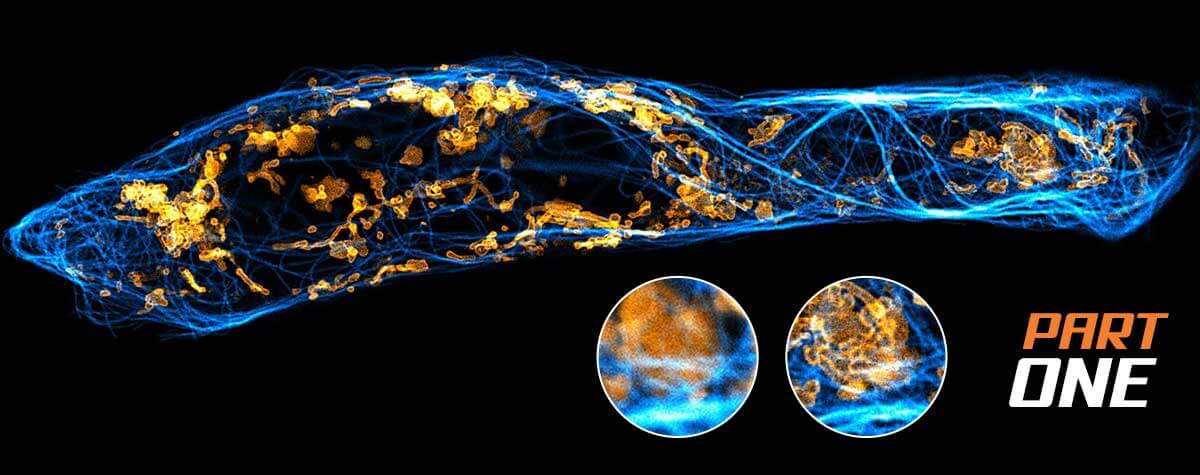
MATRIX STED –
MATRIX STED is the next level of STED microscopy – combining superior resolution with outstanding signal quality and clarity.
many eyes see more than one
Fluorescence microscopy is widely used in research, diagnostics, and biomedical imaging. It enables the non-invasive multi-dimensional (X, Y, Z, t) visualization of structures and molecules and is ideally suited for the examination of medical and biological samples. Unfortunately, the resolution of conventional light microscopes is limited by diffraction to about half the wavelength of light (Abbe, 1873). In the last few decades, numerous technological advances such as novel light sources, point detectors, beam scanners and several levels of automatization have been implemented in light microscopes. A fundamental step was the advent of super resolution fluorescence microscopy techniques, which offer resolution capabilities beyond the diffraction-limit. In particular, stimulated emission depletion (STED) microscopy (Hell and Wichmann, 1994) routinely resolves structures as small as 20 nm, in a wide variety of sample types, including living cells where the visualization of cellular dynamics can offer important biological insights.
Superresolution microscopy performs best in regions of the sample that are relatively thin, such that there is minimal out-of-focus background signal to degrade the quality of the final image. For the same reason, thicker samples can prove challenging to image via superresolution microscopy, due to low signal-to-back-ground ratios. The MATRIX detector addresses this limitation by eliminating out-of-focus background, therefore enabling low-background 2D and 3D STED imaging, even in thick samples, with dense labeling and overlapping structures.
The challenge: bright high-resolution images with background suppression
An ideal fluorescence microscope combines many functionalities and key requirements. First, a microscope must acquire bright fluorescent images. For this, the microscope must be optimized to collect as many photons from the fluorophores in the sample as possible. Second, a good microscope should have strong optical sectioning, i.e., acquire signals from an isolated single plane only. In the 1950s, this lead Minsky to the invention of the confocal microscope (Minsky, 1957). However, in practice, not only light from out-of-focus planes is rejected by the pinhole but as well parts of the light from the focal plane (Egner et al., 2020). It is important to note that this is not a binary action. The fraction of light being recorded decreases with the square of the distance of the source from the focal plane. Therefore, for thick samples, many layers of background can still sum up and contribute a significant amount of unwanted signal.
The third requirement of a good microscope is high resolution. Several concepts have been proposed for computationally improving the spatial information of confocal images (I. J. Cox and C. J. R. Sheppard, 1983), but these strategies are ultimately limited by the available signal-to-noise ratios (SNR) and the signal to background ratio in the images (SBR) (Sheppard et al., 1992). Theoretically, a resolution improvement of √2 over a 1-airy-unit-pinhole can also be achieved using a very small pinhole (Schrader et al., 1996; Wilson, 1990). However, this resolution increase is counterbalanced by a strong reduction of detected light through the pinhole, which decreases SNR and image quality. In practice, these methods have their merits, but the true breaking of the diffraction barrier came only with the advent of super resolution microscopy methods, including STED microscopy.
The increase in resolution offered by STED microscopy brings new challenges along with it. In a conventional microscope, molecules within the same focal volume emit fluorescence simultaneously and cannot be separated due to lack of resolution. With increasing resolving power, and therefore a decreasing focal volume, fewer and fewer molecules are emitting fluorescence simultaneously. After all, the whole point of super resolution is to look at small structures separately. Consequently, the perceived signal level will drop, but only because fewer molecules are fluorescing at any given time. Nevertheless, the STED effect only acts on fluorophores in the focal plane while fluorophores in other layers are not de-excited. This results in signal being recorded from the focal plane with improved resolution but reduced intensity, whereas background signal emanating from the entire thickness of the sample contributes with full intensity. As a result, the SBR is typically reduced in STED images of thick samples, sometimes to the point where resolution must be sacrificed to avoid drowning in-focus signal.
The solution: MATRIX STED
The MATRIX detector is the key component of MATRIX STED, a state-of-the-art 3D STED microscope with superior background reduction. This detector relies on the principle that “many eyes see more than one” (Box 1). It consists of multiple avalanche photo diode (APDs) elements, each with an extraordinarily high quantum efficiency (>50% at 500 nm), arranged in a hexagonal arrangement on a single detector chip (Fig. 1). In contrast to conventional confocal and STED microscopy, MATRIX imaging is performed with an open pinhole so that a maximal amount of light from the sample is collected. Each of the elements records a part of the point spread function (PSF) – approximately 0.3 AU per individual MATRIX element – thereby enabling improved characterization of the sample.
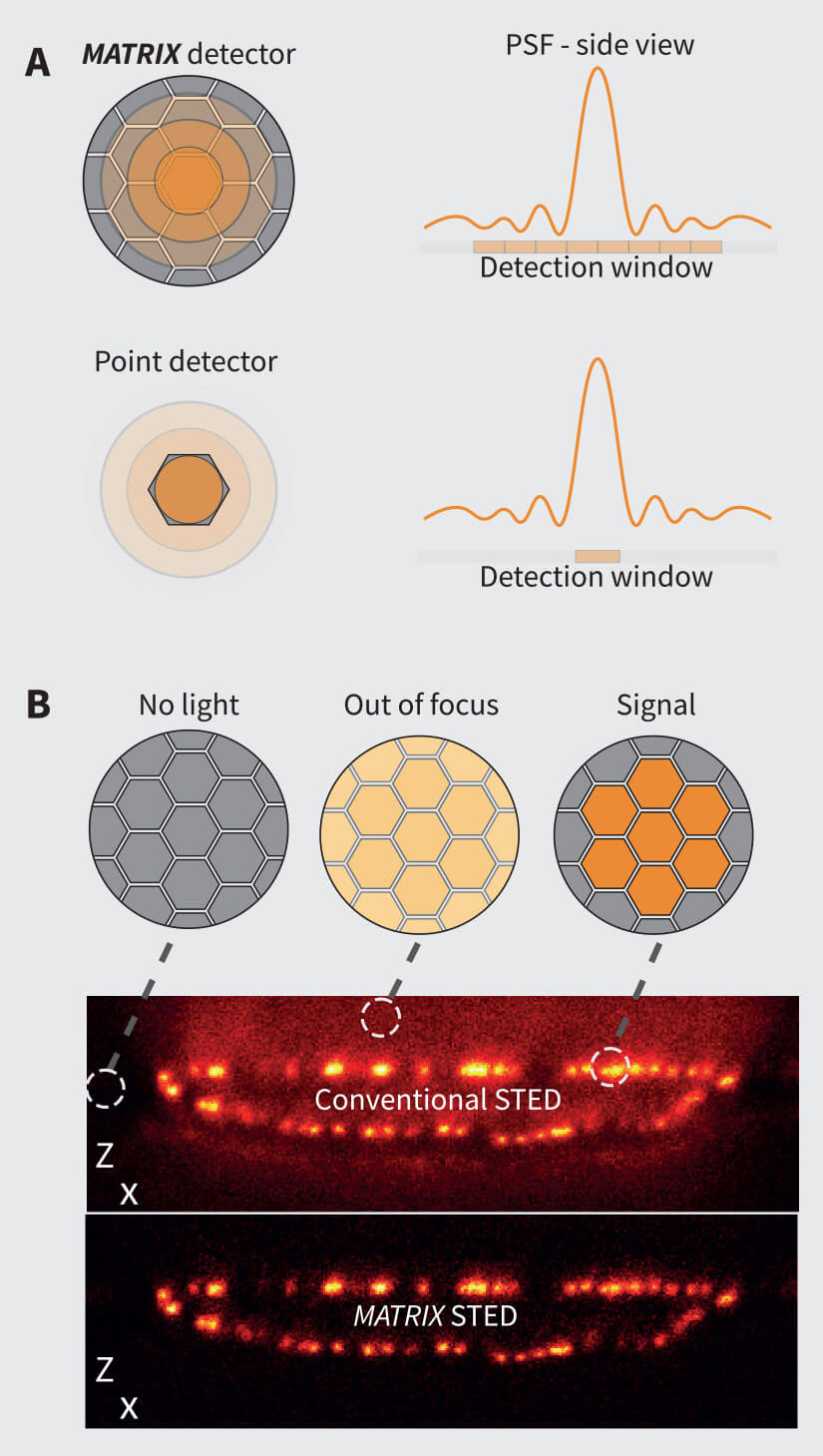
Figure 1. The MATRIX Principle. (A) The MATRIX detector consists of more than 20 individual elements that allow the detection of signal within the focal plane together with signal from planes above or below the structure of interest. The many individual elements of the MATRIX create a large detector array that records the centre of the PSF as well as its lateral parts. Compared to the single point detector, much more information about the PSF is recorded. (B) The working principle of the MATRIX detector is the detection and separation of out-of-focus background that emerges from structures above and below the current focal plane. In contrast to the pinhole that can only limit the stray light from relatively far apart structures, the MATRIX enables the removal of stray light from nearby structures as they occur in dense regions of the sample.
The additional information content of the MATRIX images can be combined with several powerful post-processing algorithms to improve the quality and clarity of the final image. For example:
1. Differential Detection:
Data acquired using the MATRIX detector can be efficiently used to differentiate between in-focus and out-of-focus contributions of the sample. With this information, outof- focus background can be removed from the acquired images.
2. Deconvolution:
In contrast to images from confocal microscopes with single point detectors, images acquired with a MATRIX detector contain a large amount of additional information. In particular, the amount of signal from the focal plane in comparison to out-of-focus contributions is known. This additional information results in greatly improved results when further post-processing steps such as deconvolution are applied.
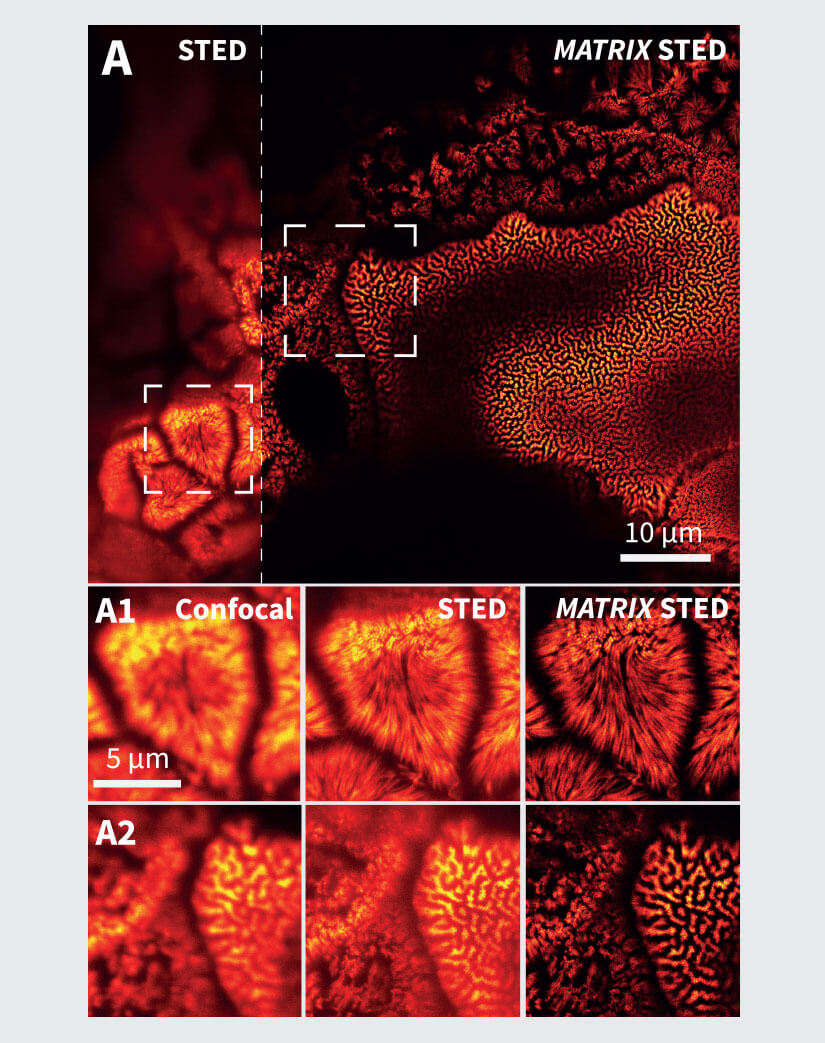
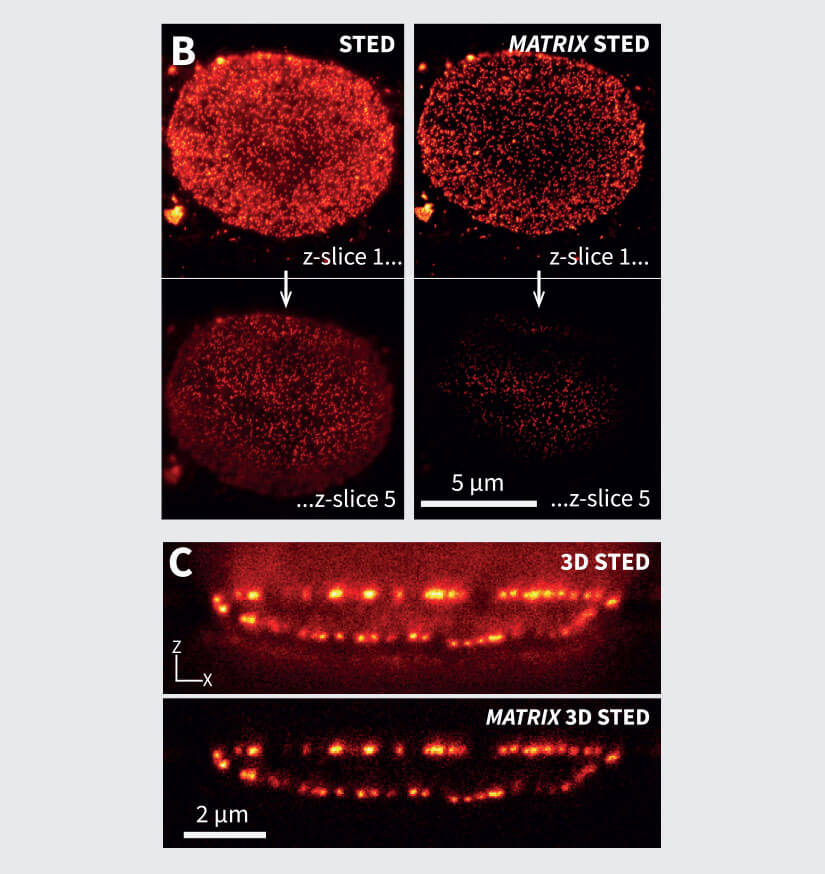
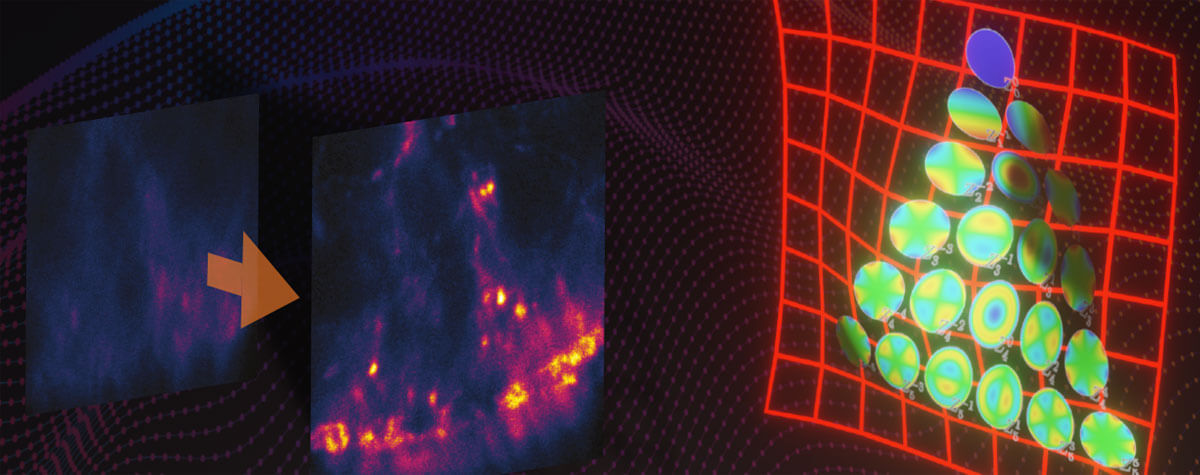
Figure 2 MATRIX STED improves STED clarity and quality. (A) Improved signal clarity in a 3D intestinal epithelial cell culture model. Comparing Confocal, STED and MATRIX STED (A1-A2) reveals improved signal to background ratio by removing out-of-focus blur through MATRIX postprocessing. Caco-2 cells labelled with Phalloidin-abberior STAR RED. (B) Optical sectioning is clearly improved in volumes acquired with STED. MATRIX postprocessing enables removal of structures that are not exactly in the focal plane thereby advancing optical sectioning by MATRIX postprocessing. Shown are STED stacks of nuclear pore complexes in mammalian cells (C) The improved z-sectioning is a key advantage in 3D STED volume imaging as the reduction of out-of-focus blur enables improved 3D separation and 3D rendering with increased signal clarity. 3D STED Z-scan of nuclear pore complex proteins in mammalian cells. Images were acquired with comparable pinhole size of approx. 0.7AU. Immunolabelling was performed with abberior STAR RED.
By starting with a clearer and better image compared to microscopes with conventional point detectors, microscopes equipped with MATRIX detectors offer users an edge in a variety of image analysis pipelines, including improved separation of densely packed objects, improved particle counting, size estimation, intensity measurements, smoothing, and deconvolution. Although background removal can in theory also be achieved by deconvolution alone, there are two requirements that are hard to achieve in practice: First, deconvolution algorithms require very accurate knowledge about the shape of the point-spread function (PSF) far away from the focus. Tiny deviations caused by aberrations or inhomogeneities of the sample can have large effects that can lead to inaccurate results. Second, deconvolution requires the recording of 3D-stacks over the full thickness of the sample or at least the full thickness from which there is a background contribution. This considerably slows down acquisition and increases bleaching and phototoxicity. With MATRIX STED, the background contributions can be directly read out and removed from using only a single recorded xy-plane, without detailed knowledge about the actual PSF.
Gedankenexperiment on the properties of the MATRIX Detector

This simple experiment exemplifies the advantage of having many detector elements as in the matrix detector compared to only a single detector: (1) Extend your arm in front of you and stick out the thumb of your hand as in the illustration on the left. (2) Focus on your thumb with one eye (while the other eye is closed). (3) Switch eyes, alternating between open and closed. When switching between eyes, your thumb will seem to jump sideways in front of whatever is in the background. This demonstrates that having two eyes (two detectors) allows you to distinguish foreground from background easily. The simple fact that your thumb moves in front of the background is enough to tell the two apart. Similarly, the over twenty “eyes” (detectors) of the MATRIX can be used to determine the amount of background for each pixel and remove it.
MATRIX STED in biomedical applications
In biology, regions of interest are often densely packed areas within cells and tissues, as it is these areas with the densest crowding of organelles or proteins that are the sites of high biological activity. At low-density areas such as the outer part of the cell where organelles and filaments are often not overlapping, superresolution microscopy excels and allows to resolve and discriminate structures very well. However, areas with a high density of labeled structures remain challenging as the contribution of fluorescence from neighboring structures (in 3D) makes it hard to separate individual structures unambiguously (Fig. 2).
The background reduction offered by MATRIX STED is advantageous for such applications and sample types, where crowded membranes and organelles can generate background haze when imaged via conventional STED microscopy.
For example, we show in Fig. 2A the conventional vs. MATRIX STED imaging of Caco-2 epithelial cells labeled for actin. These polarized cells are grown on a paper matrix and are characterized by many densely packed cell protrusions called microvilli. With MATRIX STED, it is possible to greatly improve separation of microvilli from the background (Fig. 2 A1-A2) enabling improved 3D visualization compared to conventional STED (for further examples and 3D movies, please visit www.abberior.com). Also, when imaging proteins in the nuclear membrane as shown in Fig. 2B for nuclear pore complex proteins, MATRIX STED removes fluorescent contributions from nuclear pores that are not in the current focal plane, therefore enhancing the z-sectioning capability of 2D STED far beyond what is possible with a pinhole. The effect of background removal and enhanced separation in z is also evident with 3D STED (Fig. 2C). All in all, the removal of outof- focus light using MATRIX detection reduces the spillover of out-of-focus structures in each individual z-plane and improves the separation of densely packed and labeled objects such as cytoskeletal elements or mitochondria in the vicinity of the nucleus.
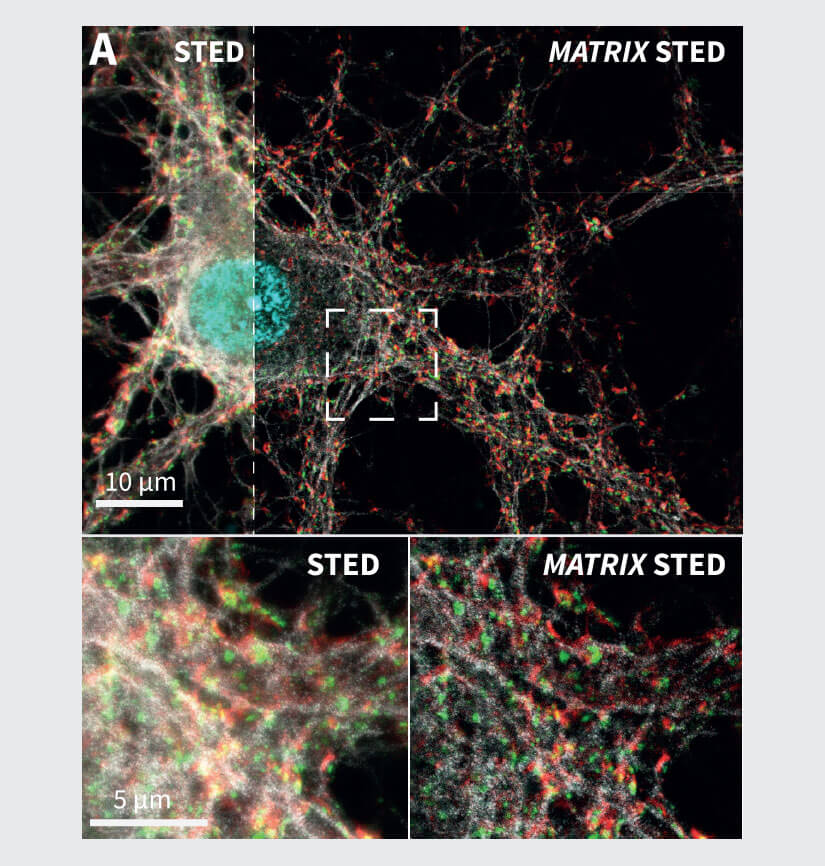
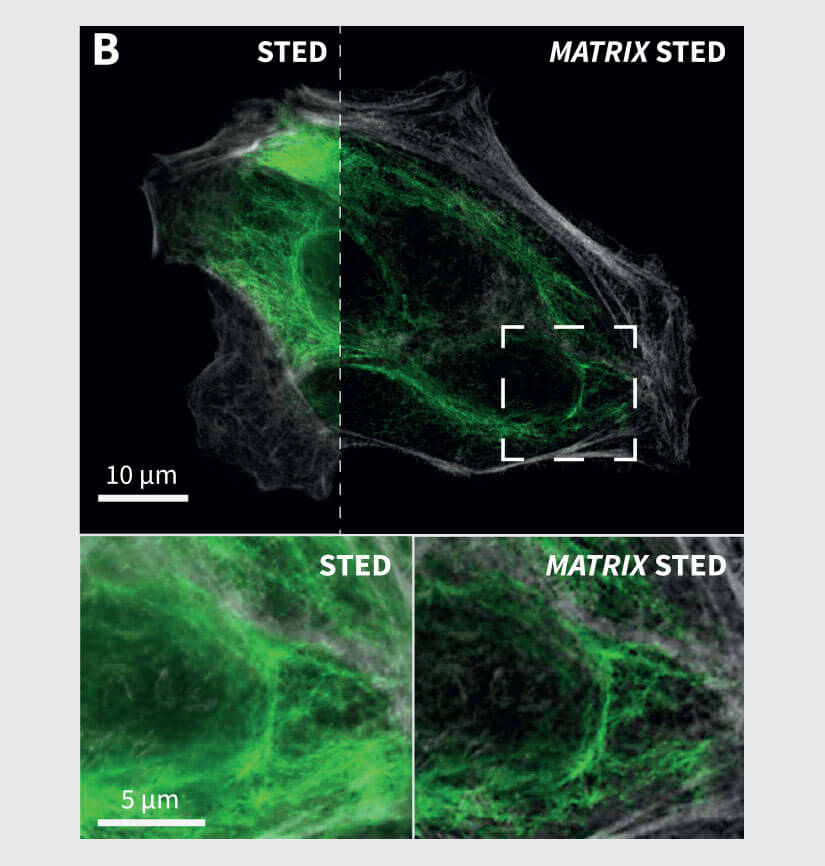
Figure 3 Application examples from neurobiology and cell biology. (A) MATRIX STED of primary neurons labelled for Spectrin (gray), Bassoon (green), Dapi (cyan) and actin (phalloidin, red). (B) MATRIX STED of Vimentin (green) and actin (phalloidin, grey) in mammalian cells demonstrate the advantages of MATRIX detection for imaging densely packed structures like cytoskeletal elements, e.g. vimentin or phalloidin. MATRIX detection enables improved separation of individual filaments, zsectioning and therefore selective optical de-crowding of tightly packed areas in the cell. Labelling: Neurons (A): Primary antibody against Bassoon and Spectrin labelled with secondary antibodies with abberior STAR RED and abberior STAR ORANGE, Phalloidin was directly coupled to abberior STAR GREEN. (B): Mammalian Cells with Vimentin and Phalloidin labelled with secondary antibodies with abberior STAR RED and abberior STAR 580.
Another application for MATRIX STED is the separation of protein complexes in synapses shown in Fig. 3A, where the synaptic protein Bassoon is imaged together with actin to visualize synaptic boutons. The spectrin labeling additionally highlights the cytoskeleton of the neuronal processes. The selective removal of background via the MATRIX detector improves the separation of these structures while also achieving significantly higher in-focus signal levels compared to a single point detector image. Also, for the labeling of several cytoskeletal elements such as vimentin and actin, the increased clarity and separation of filaments allows for improved analysis and quantification especially in areas where many filaments overlap (Fig 3B). In general, this is possible for all structures with high background such as tissue slices or tightly packed cell aggregates.
Summary
The MATRIX detector removes background and increases optical sectioning. Combining one or two MATRIX detectors with spectral RAINBOW detection yields excellent STED image quality for densely packed samples and allows the user to explore a large variety of dyes and experimental conditions, including fixed-cell, live-cell and tissue imaging.
References
Abbe, E., 1873. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Archiv f. mikrosk. Anatomie 9, 413–468. https://doi.org/10.1007/BF02956173
Egner, A., Geisler, C., Siegmund, R., 2020. STED Nanoscopy, in: Salditt, T., Egner, A., Luke, D.R. (Eds.), Nanoscale Photonic Imaging, Topics in Applied Physics. Springer International Publishing, Cham, pp. 3–34. https://doi.org/10.1007/978-3-030-34413-9_1
Hell, S.W., Wichmann, J., 1994. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780. https://doi.org/10.1364/ OL.19.000780
I. J. Cox and C. J. R. Sheppard, 1983. Scanning optical microscope incorporating a digital framestore and microcomputer. Applied Optics, Vol. 22, Issue 10, pp. 1474-1478. https://doi.org/10.1364/ AO.22.001474
Minsky, M., 1957. MICROSCOPY APPARATUS. US Patent US3013467A.
Schrader, M., Hell, S.W., van der Voort, H.T.M., 1996. Potential of confocal microscopes to resolve in the 50–100 nm range. Appl. Phys. Lett. 69, 3644–3646. https://doi.org/10.1063/1.117010
Sheppard, C.J.R., Gu, M., Roy, M., 1992. Signal-to-noise ratio in confocal microscope systems. Journal of Microscopy 168, 209–218. https://doi.org/10.1111/j.1365-2818.1992.tb03264.x
Wilson, T. (Ed.), 1990. Confocal microscopy. Acad. Press, London